Clariscience “Dispositivi paziente-specifici: nuovi vantaggi, ma anche nuovi rischi?”

I progressi della tecnologia nel settore sanitario, nella scienza dei materiali, nelle tecnologie di imaging e di produzione hanno reso possibile la creazione di dispositivi medici personalizzati, realizzati per rispondere alle esigenze e alle caratteristiche specifiche di un particolare paziente. Nell’ottica di garantire ai pazienti un livello ottimale di sicurezza e prestazioni anche per questo tipo di dispositivi, l’International Medical Device Regulators Forum (IMDRF) ha pubblicato dei documenti di orientamento che forniscono indicazioni armonizzate a livello internazionale per definirne le diverse categorie e l’inquadramento regolatorio.
La linea guida IMDRF N.49 Definitions for Personalized Medical Devices distingue i dispositivi medici personalizzati in:
- I dispositivi medici su misura (custom made), specificatamente fabbricati sulla base di una prescrizione da parte di una figura professionale autorizzata e destinati a uno specifico paziente.
- i dispositivi prodotti in serie o in lotti omogenei mediante un processo che può essere convalidato e riprodotto, che possono essere:
- dispositivi medici paziente-specifici (patient-matched): realizzati da un fabbricante per un determinato individuo, all’interno di determinato “range progettuale”.
- dispositivi medici adattabili: dispositivi che, una volta fabbricati, devono essere regolati, assemblati o modellati da un operatore sanitario presso il “point of care” per renderli adatti alle specifiche esigenze del paziente, seguendo le istruzioni fornite dal fabbricante.
Questo articolo, a cura di Clariscience, approfondisce i requisiti dei dispositivi medici paziente-specifici (patient-matched). Un dispositivo paziente-specifico è un dispositivo medico che, sebbene possa essere progettato con la consulenza di un professionista sanitario, viene prodotto sotto la responsabilità del fabbricante, mediante un processo che può essere convalidato e riprodotto in serie o in lotti omogenei. Tali dispositivi sono realizzati in modo da essere adatti (in inglese: matched) all’anatomia di un determinato paziente, ma all’interno di un determinato range progettuale.
Esempi di dispositivi paziente-specifici sono:
- piastre per la fissazione di fratture ossee realizzate mediante stampa 3D, sulla base di un modello e di file/immagini del paziente
- guide chirurgiche (es. per l’artroplastica del ginocchio) o per il posizionamento di viti, realizzate mediante stampa 3D sulla base di dati di imaging (MR o TC) per un paziente specifico
- impianti mandibolari prodotti con la stampa 3D, a partire da immagini digitali
- lenti a contatto su ordinazione
- ortesi da indossare esternamente, basate su immagini 3D
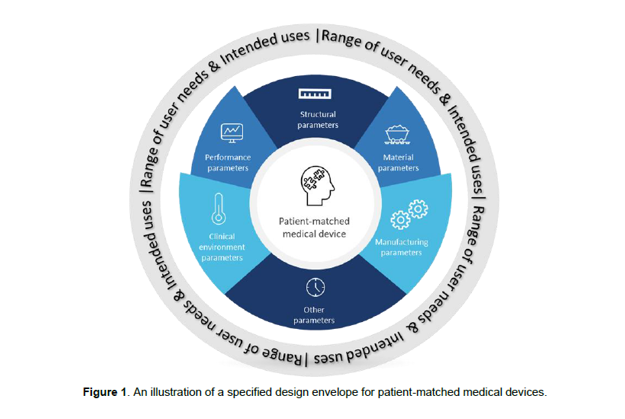
La linea guida IMDRF N.49 introduce il concetto di “range progettuale” (specified design envelope) per i dispositivi paziente-specifici, che viene sviluppato nella linea guida IMDRF N.74 Personalized Medical Devices – Production Verification and Validation: il “range progettuale” è un intervallo di parametri definiti dal fabbricante in modo da garantire che qualsiasi dispositivo prodotto all’interno di questo range rispetti i requisiti di sicurezza e prestazione previsti.
Per definire un “range progettuale”, il fabbricante deve identificare in modo inequivocabile tutti i parametri rilevanti che caratterizzano il design del dispositivo e stabilire esplicitamente gli intervalli di riferimento per ciascun parametro.
Le categorie di parametri, per i quali devono essere stabiliti dei limiti espliciti sono:
- parametri strutturali: dimensioni, aree, volumi, forme, angoli, posizioni relative, distanze consentite e altri parametri geometrici del dispositivo
- proprietà dei materiali: le caratteristiche biologiche, fisiche, chimiche delle materie prime utilizzate nella produzione e la conformità agli standard di riferimento, considerando anche eventuali riutilizzi del materiale
- parametri di fabbricazione: devono essere identificati tutti i parametri che possono essere variati durante le fasi di produzione, post-produzione, assemblaggio, pulizia, sterilizzazione, all’imballaggio ed etichettature e devono essere stabiliti limiti espliciti per ciascuno di essi
- parametri relativi al contesto clinico: limiti correlati al contesto clinico per il quale il dispositivo è destinato ad essere utilizzato
- parametri di prestazione: parametri relativi alle prestazioni del dispositivo quando utilizzato secondo la sua destinazione d’uso
- altri
Se il workflow per la realizzazione del dispositivo prevede l’utilizzo di dati da immagini, ottenute mediante ad es. tomografia computerizzata, risonanza magnetica, ultrasuoni, il fabbricante deve considerare anche i fattori relativi all’acquisizione dei dati e all’elaborazione delle immagini.
I limiti delle variabili progettuali devono essere definiti nell’ambito del processo di analisi dei rischi del fabbricante, nel quale dovrà essere considerato uno scenario rappresentativo del caso peggiore (“worst case”) nell’ambito della destinazione d’uso prevista del dispositivo.
Le attività di verifica e validazione della progettazione dovranno dimostrare che il processo di fabbricazione è controllato e riproducibile, per cui qualsiasi dispositivo prodotto all’interno del range progettuale dovrà essere conforme alle esigenze dell’utilizzatore e all’uso inteso previsto. La validazione della progettazione dovrà essere completata con una valutazione clinica.
Dal punto di vista regolatorio, per poter essere immessi sul mercato, i dispositivi medici paziente-specifici sono considerati come i dispositivi prodotti in serie: devono essere quindi correttamente classificati dal fabbricante e devono rispettare tutti i requisiti pre- e post- market previsti dal Regolamento 2017/745, in base alla loro classe di rischio, come anche richiamato, in particolare, al paragrafo 7 della linea guida IMDRF N.58 Personalized Medical Devices – Regulatory Pathways: il fabbricante dovrà quindi assumersi l’onere di dimostrare la conformità dei dispositivi ai requisiti di sicurezza e prestazione applicabili, compresi quelli di prestazione clinica, di etichettatura, di sorveglianza post-commercializzazione, la capacità di intraprendere azioni correttive, e di rispettare gli obblighi di vigilanza.
Sebbene il frame concettuale alla base della dimostrazione della conformità dei dispositivi medici paziente-specifici sia quello applicabile ai dispositivi medici in genere, le loro particolari caratteristiche, delineate in questo articolo, possono rendere utile richiedere l’affiancamento di un professionista esperto per l’interpretazione dei requisiti applicabili e per l’assistenza nella preparazione della relativa documentazione tecnica.