Clariscience: L’usabilità vista dalla FDA

Per poter commercializzare un dispositivo medico negli Stati Uniti è necessario dimostrare non solo che questo sia sicuro ed efficace, ma anche che possa essere utilizzato, nell’ambito della destinazione d’uso prevista, senza incorrere in errori d’uso che possano comportare rischi inaccettabili per i pazienti o per gli operatori.
Per assistere i fabbricanti nella progettazione dei dispositivi medici, la US Food and Drug Administration ha da tempo pubblicato la guida Applying Human Factors and Usability Engineering to Medical Devices – Guidance for Industry and FDA Staff, che descrive le attività che i fabbricanti dovrebbero svolgere per una progettazione efficace dell’interfaccia utente di un dispositivo medico applicando l’ingegneria dei fattori umani, in modo da eliminare o ridurre il più possibile gli errori di utilizzo che si verificano durante l’uso del dispositivo e che potrebbero causare danni o compromettere il trattamento .
L’interfaccia utente di un dispositivo è costituita da tutti gli elementi attraverso i quali l’utilizzatore interagisce con il dispositivo (la zona rossa in figura 1), sia nelle fasi preliminari all’uso (ad esempio, disimballaggio, impostazione, calibrazione), sia durante il suo impiego, sia nell’esecuzione di interventi di manutenzione (ad esempio, pulizia, sostituzione della batteria, riparazione).
Figura 1
(immagine da: Applying Human Factors and Usability Engineering to Medical Devices
– Guidance for Industry and FDA Staff
Il dispositivo riceve l’input dall’utilizzatore, elabora i dati e fornisce un feedback. L’utilizzatore percepisce, interpreta ed elabora le informazioni fornite dal dispositivo, prende delle decisioni e può avviare ulteriori cicli di interazione.
Studiare come l’interfaccia utente influisce sulle interazioni che le persone hanno con il dispositivo, e quindi con la tecnologia impiegata, è l’obiettivo dell’ingegneria dei fattori umani (Human Factor Engineering, HFE), chiamata anche dell’ingegneria dell’usabilità (Usability Engineering, UE), terminologia utilizzata nella norma IEC 62366 1:2015 Medical devices – Part 1: Application of usability engineering to medical devices, che è uno degli standard riconosciuti dall’FDA.
L’FDA prevede che l’ingegneria dei fattori umani debba essere utilizzata nella progettazione e nella gestione dei rischi del dispositivo medico.
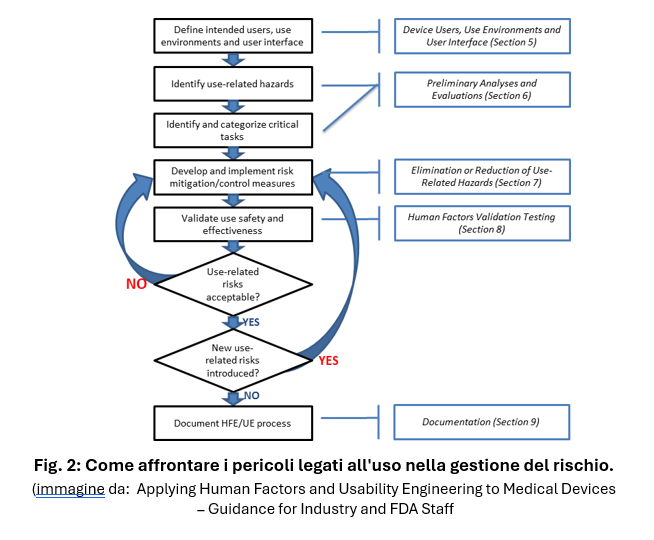
La guida Applying Human Factors and Usability Engineering to Medical Devices individua tre step per una valutazione HFE efficace:
- L’identificazione dei rischi correlati all’uso del dispositivo (use-related hazards), e delle situazioni d’uso pericolose;
- Lo sviluppo e l’applicazione di misure per eliminare o ridurre i rischi correlati all’uso del dispositivo che potrebbero causare danni al paziente o all’operatore.
- La dimostrazione mediante test di validazione (HFE validation) che il design finale dell’interfaccia utente del dispositivo supporta un uso sicuro ed efficace.
 Nello sviluppo dei dispositivi medici i tre principali aspetti che devono essere caratterizzati e documentati sono:
Nello sviluppo dei dispositivi medici i tre principali aspetti che devono essere caratterizzati e documentati sono:
- l’utilizzatore previsto,
- l’ambiente di utilizzo,
- le interfacce utente del dispositivo.
Nella Current Good Manufacturing Practice (cGMP, Quality System Regulation Preamble to Final Rule 21 CFR Parts 808, 812, and 820) viene affermato che i fattori umani devono essere presi in considerazione e documentati fin dalle prime fasi del processo di progettazione.
In particolare, deve essere eseguita un’analisi preliminare per identificare le azioni che devono essere compiute dall’utilizzatore (user task), le componenti dell’interfaccia e i possibili problemi di utilizzo, con lo scopo di classificare tutti i task in base alla gravità delle possibili conseguenze di un errore di esecuzione e redigere un elenco dei task considerati “critici”, ovvero quelle azioni che, se eseguite in modo errato o non eseguite affatto, potrebbero causare danni gravi.
L’identificazione dei task critici è il risultato di un’analisi del rischio definita use-related risk analysis (URRA) che può essere svolta utilizzando diverse metodologie, quali FMEA, analisi dei task, analisi dei problemi già noti, valutazioni euristiche, valutazione di esperti.
I task critici possono essere identificati anche mediante un approccio empirico, con la valutazione di dispositivi simili, o mediante una valutazione formativa che preveda test di uso simulato. È importante che, una volta identificati, tutti i rischi vengano eliminati o mitigati il più possibile, prima di avviare il test di validazione finale dell’usabilità.
I test di validazione dei fattori umani sono generalmente condotti in condizioni di uso simulato, ma quando necessario, i dati possono essere raccolti anche in condizioni di uso reale o come parte di uno studio clinico. È fondamentale che i partecipanti ai test di validazione rappresentino la popolazione degli utenti previsti. Il numero necessario di partecipanti al test dovrebbe essere determinato in base ai risultati delle analisi e delle valutazioni preliminari ma, in generale, non dovrebbe essere inferiore a 15 persone.
Affinché i risultati dei test di validazione dei fattori umani dimostrino un uso sicuro ed efficace da parte degli utilizzatori statunitensi, i partecipanti ai test devono risiedere negli Stati Uniti. Gli studi condotti in altri Paesi o con persone non residenti negli Stati Uniti possono essere influenzati (positivamente o negativamente) dalle diverse pratiche cliniche esistenti in altri Paesi.
I risultati del test di validazione sono descritti in un Report, che deve essere redatto secondo la struttura descritta nell’Appendice A della guida; non è necessario che vengano inclusi tutti i dati del test, ma deve essere descritto come tutti i rischi correlati all’uso del dispositivo siano stati identificati, valutati e mitigati.
Nel Dicembre 2022 la US Food and Drug Administration (FDA) ha pubblicato un documento dal titolo: Content of Human Factors Information in Medical Device Marketing Submissions, ancora in fase di draft, per chiarire ai fabbricanti quali informazioni sulla valutazione dei fattori umani è necessario includere nelle richieste di autorizzazione alla commercializzazione (premarket submissions) dei dispositivi medici in USA.
Le richieste vengono classificate in categorie “HF Submission Category” in funzione del rischio associato ai dispositivi e le informazioni che devono essere fornite in base alla categoria sono riportate nella tabella seguente:
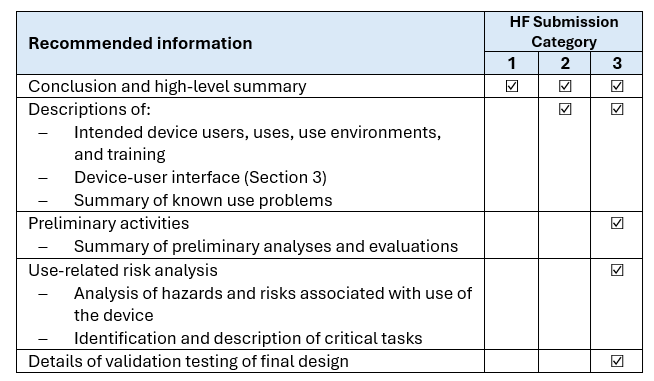
Pur essendo disponibili documenti di orientamento pubblicati dall’autorità regolatoria, data la complessità dell’argomento può essere utile avvalersi di un consulente esperto per applicare il processo di ingegneria dei fattori umani nella progettazione e nella gestione dei rischi del dispositivo medico e per prepararsi a commercializzare con successo i propri dispositivi negli Stati Uniti.